jvarkit
Bam2Raster

BAM to raster graphics
Usage
This program is now part of the main jvarkit tool. See jvarkit for compiling.
Usage: java -jar dist/jvarkit.jar bam2raster [options] Files
Usage: bam2raster [options] Files
Options:
-clip, --clip
Show clipping
Default: false
-depth, --depth
Depth track height.
Default: 100
--groupby
Group Reads by. Data partitioning using the SAM Read Group (see
https://gatkforums.broadinstitute.org/gatk/discussion/6472/ ) . It can
be any combination of sample, library....
Default: sample
Possible Values: [readgroup, sample, library, platform, center, sample_by_platform, sample_by_center, sample_by_platform_by_center, any]
-h, --help
print help and exit
--helpFormat
What kind of help. One of [usage,markdown,xml].
--highlight
hightligth those positions.
Default: []
--mapqopacity
How to handle the MAPQ/ opacity of the reads. all_opaque: no opacity,
handler 1: transparency under MAPQ=60
Default: handler1
Possible Values: [all_opaque, handler1]
--limit, --maxrows
Limit number of rows to 'N' lines. negative: no limit.
Default: -1
-minh, --minh
Min. distance between two reads.
Default: 2
-N, --name
print read name instead of base
Default: false
--noReadGradient
Do not use gradient for reads
Default: false
-nobase, --nobase
hide bases
Default: false
-o, --output
Output file. Optional . Default: stdout [20180829] filename can be also
an existing directory or a zip file, in witch case, each individual will
be saved in the zip/dir.
-R, --reference
Indexed fasta Reference file. This file must be indexed with samtools
faidx and with picard/gatk CreateSequenceDictionary or samtools dict
* -r, --region
Restrict to that region. An interval as the following syntax :
"chrom:start-end". Some jvarkit programs also allow the following syntax
: "chrom:middle+extend" or "chrom:start-end+extend" or
"chrom:start-end+extend-percent%".A program might use a Reference
sequence to fix the chromosome name (e.g: 1->chr1)
-srf, --samRecordFilter
A filter expression. Reads matching the expression will be filtered-out.
Empty String means 'filter out nothing/Accept all'. See https://github.com/lindenb/jvarkit/blob/master/src/main/resources/javacc/com/github/lindenb/jvarkit/util/bio/samfilter/SamFilterParser.jj
for a complete syntax. 'default' is 'mapqlt(1) || Duplicate() ||
FailsVendorQuality() || NotPrimaryAlignment() ||
SupplementaryAlignment()'
Default: mapqlt(1) || Duplicate() || FailsVendorQuality() || NotPrimaryAlignment() || SupplementaryAlignment()
--spaceyfeature
number of pixels between features
Default: 4
-V, --variants, --vcf
VCF files used to fill the position to hightlight with POS
Default: []
--version
print version and exit
-w, --width
Image width
Default: 1000
Keywords
- bam
- alignment
- graphics
- visualization
- png
See also in Biostars
Source code
Unit Tests
Contribute
- Issue Tracker: http://github.com/lindenb/jvarkit/issues
- Source Code: http://github.com/lindenb/jvarkit
License
The project is licensed under the MIT license.
Citing
Should you cite bam2raster ? https://github.com/mr-c/shouldacite/blob/master/should-I-cite-this-software.md
The current reference is:
http://dx.doi.org/10.6084/m9.figshare.1425030
Lindenbaum, Pierre (2015): JVarkit: java-based utilities for Bioinformatics. figshare. http://dx.doi.org/10.6084/m9.figshare.1425030
Examples
Example 1
java -jar dist/jvarkit.jar bam2raster \
-o ~/jeter.png \
-r 2:17379500-17379550 \
-R human_g1k_v37.fasta \
sample.bam
Example 2
java -jar dist/bam2raster.jar -R ref.fa -r rotavirus:150-200 data/*.bam -o out.png --limit 10 --clip --noReadGradient --highlight 175
Misc
I use the UCSC/IGV color tag ‘YC’ when available (see also samcolortag)
Screenshots
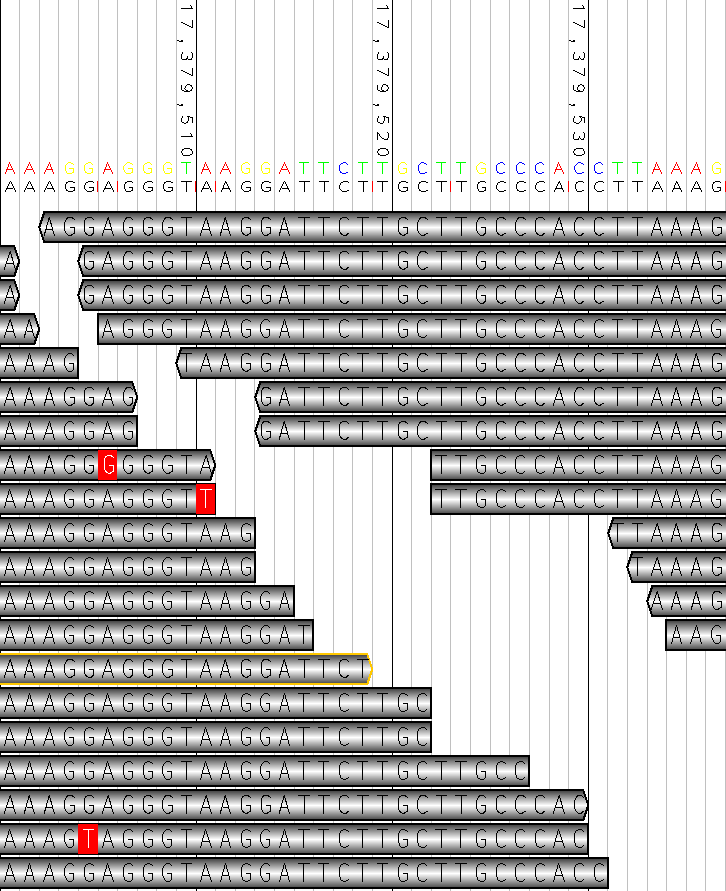

Example
$ java -jar dist/bam2raster.jar -r "scf7180000354095:168-188" \
-o pit.png \
-R scf_7180000354095.fasta scf7180000354095.bam
batch:
POS=1|123 2|345 3|456
IMAGES=
BAMS= S1|f1.bam \
S2|f2.bam \
S3|f3.bam
define run
$(1)_$(3)_$(4).png: $(2)
java -jar dist/bam2raster.jar -clip --highlight $(4) --mapqopacity handler1 --nobase -r "chr$(3):$(4)+50" --reference human_g1k_v37_prefix.fasta -o $$@ $$<
IMAGES+=$(1)_$(3)_$(4).png
endef
all: all2
$(eval $(foreach P,$(POS),$(foreach B,$(BAMS),$(call run,$(word 1, $(subst |, ,${B})),$(word 2, $(subst |, ,${B})),$(word 1, $(subst |, ,${P})),$(word 2, $(subst |, ,${P}))))))
all2: ${IMAGES}
rm -f jeter.zip
zip jeter.zip $^
screenshots


History
- 20180917 REF is now required.